News
CORRECTING SELF-INTERACTION IN SOLID STATE ELECTRONIC STRUCTURE THEORY
Image: images/NEWS/Correcting_Self_Interaction_Fig_gap_news.png
Figure caption: © Sheng Bi
Content:
|
Authors
Dr. Sheng Bi
Dr. Christian Carbogno
Prof. Dr. Igor Ying Zhang
Prof. Dr. Matthias Scheffler |
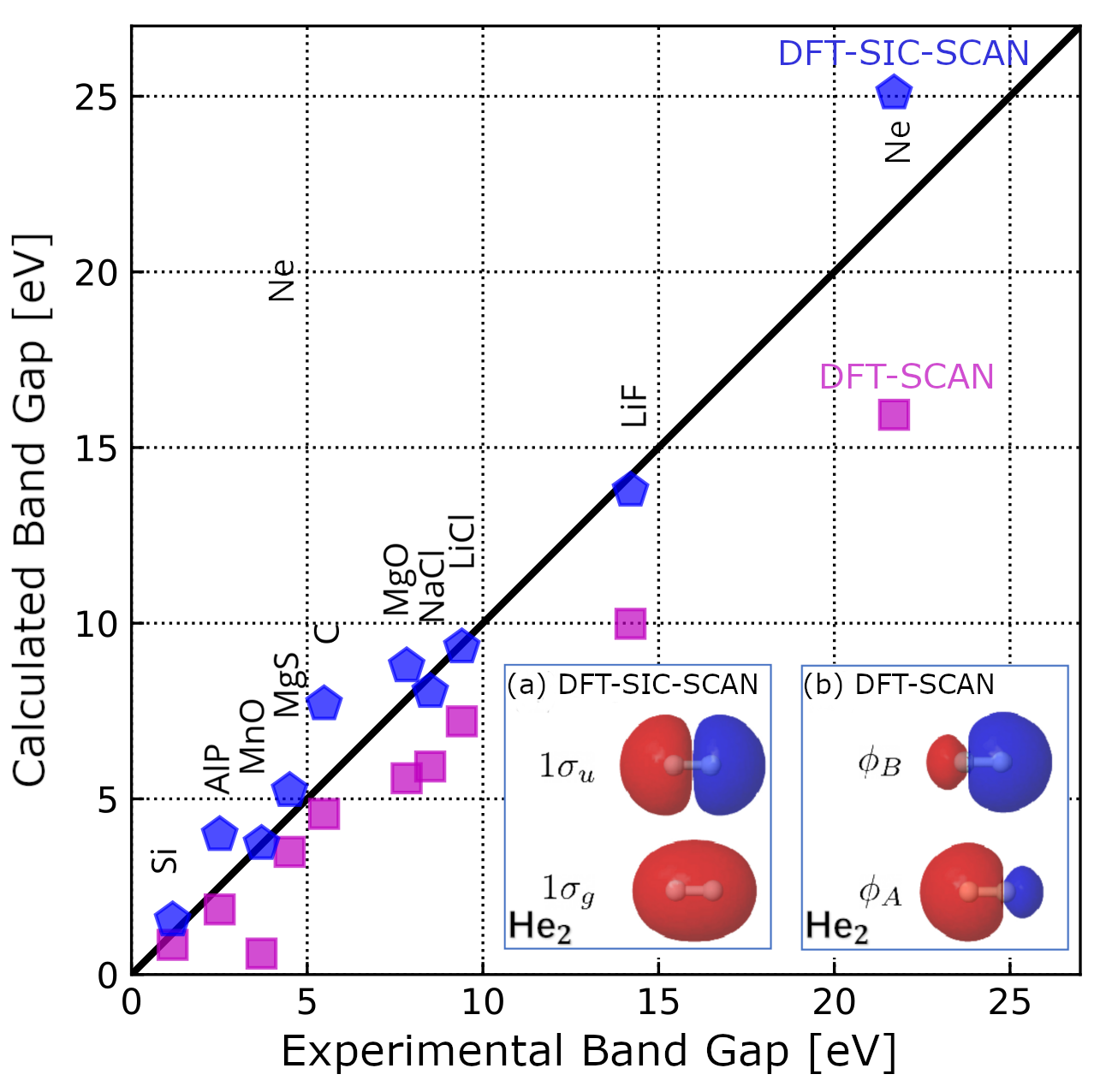
CORRECTING SELF-INTERACTION IN SOLID STATE ELECTRONIC STRUCTURE THEORYResearchers from the NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society, along with colleagues from Fudan University in Shanghai, have developed a method to correct for a well-known error in density-functional theory (DFT). This error often leads to inaccurate predictions about the electronic properties of materials, complicating the development of new materials. The so-called self-interaction error often affects the accuracy of DFT calculations. To address this issue, the team developed a new method based on the Edmiston-Ruedenberg formalism that corrects for this error and that is not affected by the numerical instabilities that have affected previously suggested correction schemes since their original proposal by Perdew and Zunger in 1980. The proposed approach significantly improves DFT predictions for key properties such as ionization potentials and band gaps in molecules and solids. For instance, the average error in band gap predictions is reduced from 31.5% to 18.5%. This is an important step in increasing the reliability of computer simulations for materials research.
Dr. Sheng Bi emphasizes that this method demonstrates how correcting the self-interaction error is key to much more accurate calculations. Although the approach is not superior in all areas, it paves the way for the development of new, improved, and hence more precise correction schemes for materials science. Read the full publication here: S. Bi, C. Carbogno, I. Y. Zhang, M. Scheffler, |




EFFICIENT DISCOVERY OF IMPROVED ENERGY MATERIALS BY A NEW AI-GUIDED WORKFLOW
Image: images/NEWS/Sobol_SHAP_Index.png
Figure caption: © Thomas Purcell, NOMAD Laboratory
Content:
Authors
Dr. Thomas Purcell
Prof. Dr. Matthias Scheffler
Dr. Luca Ghiringhelli
Dr. Christian Carbogno
|
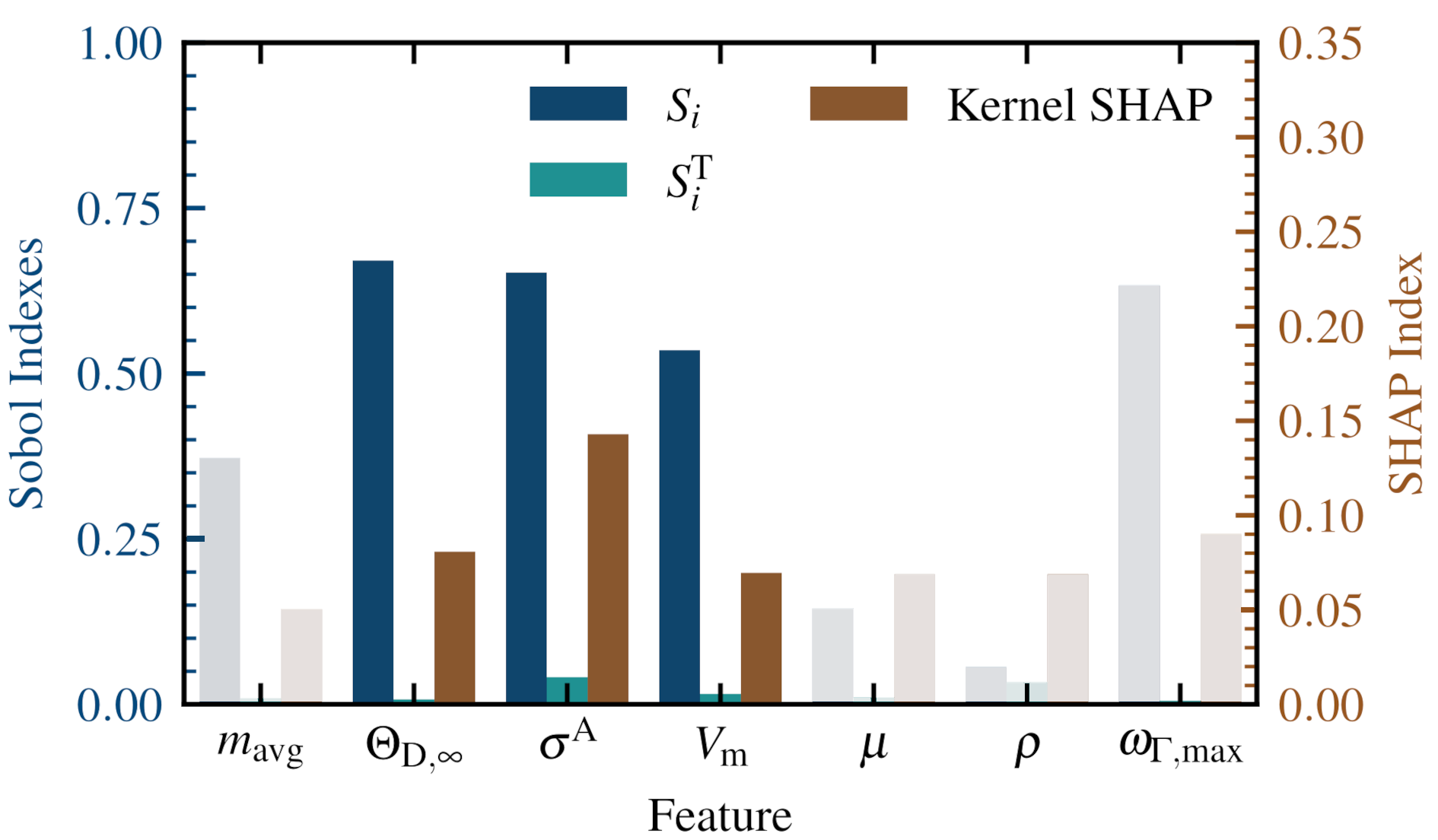
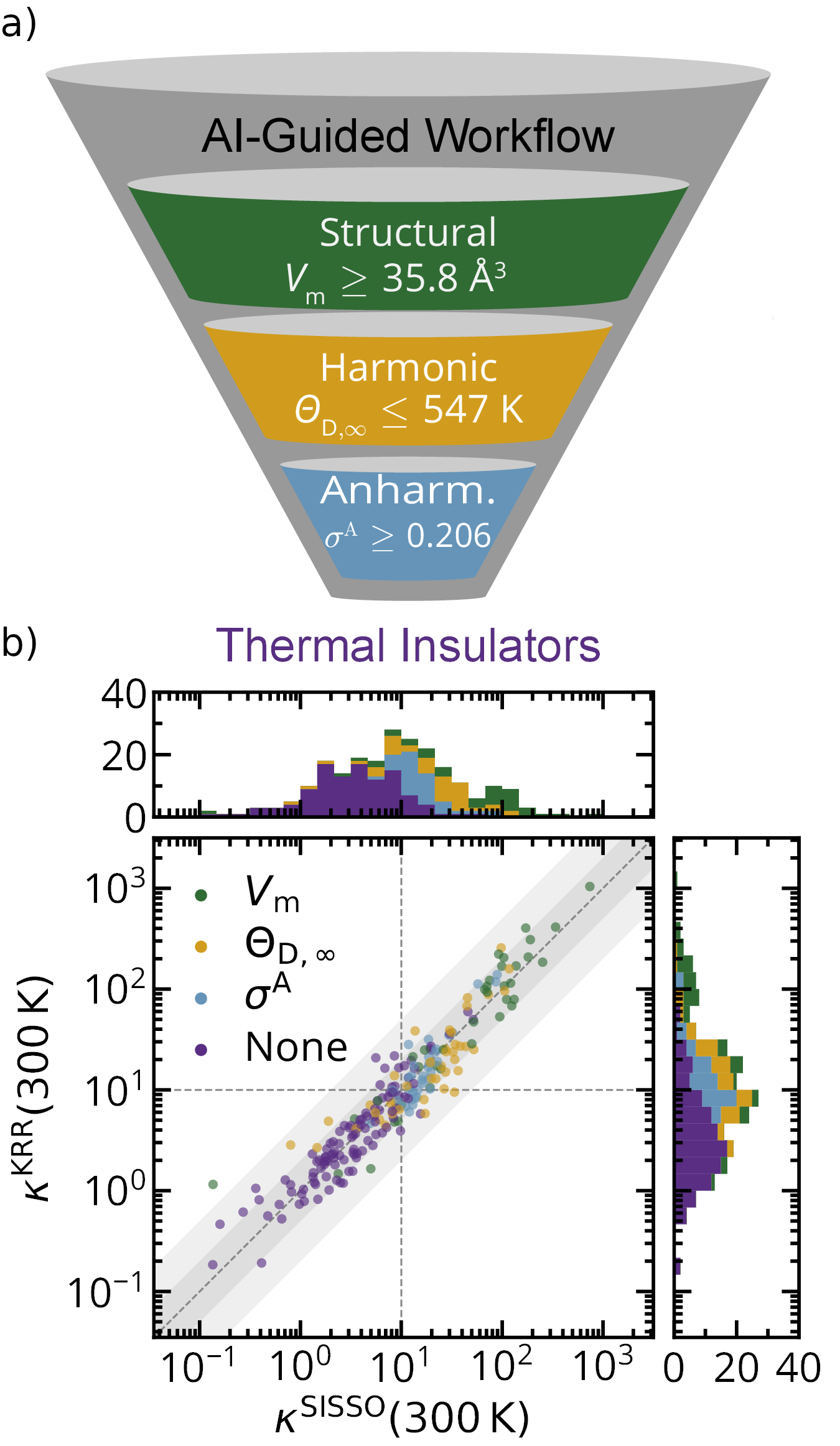
EFFICIENT DISCOVERY OF IMPROVED ENERGY MATERIALS BY A NEW AI-GUIDED WORKFLOWScientists of the NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society recently proposed a workflow that can dramatically accelerate the search for novel materials with improved properties. They demonstrated the power of the approach by identifying more than 50 strongly thermally insulating materials. These can help alleviate the ongoing energy crisis, by allowing for more efficient thermoelectric elements, i.e., devices able to convert otherwise wasted heat into useful electrical voltage. Discovering new and reliable thermoelectric materials is paramount for making use of the more than 40% of energy given off as waste heat globally and help mitigate the growing challenges of climate change. One way to increase the thermoelectric efficiency of a material is to reduce its thermal conductivity, κ, and thereby maintaining the temperature gradient needed to generate electricity. However, the cost associated with studying these properties limited the computational and experimental investigations of κ to only a minute subset of all possible materials. A team of the NOMAD Laboratory recently made efforts to reduce these costs by creating an AI-guided workflow that hierarchically screens out materials to efficiently find new and better thermal insulators. The work recently published in npj Computational Materials proposes a new way of using Artificial Intelligence (AI) to guide the high-throughput search for new materials. Instead of using physical/chemical intuition to screen out materials based on general, known or suspected trends, the new procedure learns the conditions that lead to the desired outcome with advanced AI methods. This work has the potential to quantify the search for new energy materials and increase the efficiency of these searches.
The first step in designing these workflows is to use advanced statistical and AI methods to approximate the target property of interest, κ in this case. To this end, the sure-independence screening and sparsifying operator (SISSO) approach is used. SISSO is a machine learning method that reveals the fundamental dependencies between different materials properties from a set of billions of possible expressions. Compared to other “black-box” AI models, this approach is similarly accurate, but additionally yields analytic relationships between different material properties. This allows us to apply modern feature importance metrics to shed light on which material properties are the most important. In the case of κ, these are the molar volume, Vm; the high-temperature limit Debye Temperature, θD,∞; and the anharmonicity metricfactor, σA, as illustrated in Figure 1.
Furthermore, the described statistical analysis allows to distill out rule-of-thumbs for the individual features that enable to a priori estimate the potential of material to be a thermal insulator. Working with the three most important primary features hence allowed to create AI-guided computational workflows for discovering new thermal insulators as shown in Figure 2. These workflows use state-of-the-art electronic structure programs to calculate each of the selected features. During each step materials were screened out that are unlikely to be good insulators based on their values of Vm, θD,∞, and σA. With this, it is possible to reduce the number of calculations needed to find thermally insulating materials by over two orders of magnitude. In this work, this is demonstrated by identifying 96 thermal insulators (κ < 10 Wm-1K-1) in an initial set of 732 materials. The reliability of this approach was further verified by calculating κ for 4 of these predictions with highest possible accuracy. Besides facilitating the active search for new thermoelectric materials, the formalisms proposed by the NOMAD team can be also applied to solve other urgent material science problems. |


WHEN ALL DETAILS MATTER - HEAT TRANSPORT IN ENERGY MATERIALS
Image: images/NEWS/Heat_Transport_transp_background.png
Figure caption: © Florian Knoop, NOMAD Laboratory
Content:
|
Authors
Dr. Florian Knoop
Dr. Thomas Purcell
Dr. Christian Carbogno
Prof. Dr. Matthias Scheffler |
WHEN ALL DETAILS MATTER - HEAT TRANSPORT IN ENERGY MATERIALSResearchers at the NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society have shed light of the microscopic mechanisms that determine thermal conduction in heat insulators. Powered by the advances made in the NOMAD CoE, their computational research has shown that even short-lived and microscopically localized defect structures have a substantial impact on macroscopic transport processes. This discovery could contribute to more energy-efficient technologies by allowing for the tailoring of nanoscale thermal insulators through defect engineering.
The NOMAD Laboratory researchers have recently elucidated on fundamental microscopic mechanisms that offer to tailor materials for heat insulation. This development advances the ongoing efforts to enhance energy efficiency and sustainability. The role of heat transport is crucial in various scientific and industrial applications, such as catalysis, turbine technologies, and thermoelectric heat converters that convert waste heat into electricity. Particularly in the context of energy conservation and the development of sustainable technologies, materials with high thermal insulation capabilities are of utmost importance. These materials make it possible to retain and use heat that would otherwise go to waste. Therefore, improving the design of highly insulating materials is a key research objective in enabling more energy-efficient applications. However, designing strong heat insulators is far from trivial, despite the fact that the underlying fundamental physical laws have been known for nearly a century. At a microscopic level, heat transport in semiconductors and insulators was understood in terms of the collective oscillation of the atoms around their equilibrium positions in the crystal lattice. These oscillations, called “phonons” in the field, involve zillions of atoms in solid materials and hence cover large, almost macroscopic length- and time-scales. In turn, an accurate description of the relevant dynamics across such a wide range of scales requires sophisticated methodological and computational advancements. In a recent joint publication in Physical Review B (Editors’ Suggestion) and Physical Review Letters, researchers from the NOMAD Laboratory at the Fritz Haber Institute have exploited the algorithmic and computational improvements made by the NOMAD CoE to compute thermal conductivities without experimental input at unprecedented accuracy. They demonstrated that for strong heat insulators the above-mentioned phonon picture is not appropriate. Using large-scale calculations on supercomputers at of the Max Planck Society, the North-German Supercomputing Alliance, and the Jülich Supercomputing Centre, they scanned over 465 crystalline materials, for which the thermal conductivity had not yet been measured. Besides finding 28 strong thermal insulators, six of which featured an ultra-low thermal conductivity comparable to wood, this study shed light on a hitherto typically overlooked mechanism that can be used to systematically lower thermal conductivity. “We observed the temporary formation of defect structures that massively influences the atomic motion for an extremely short period of time”, says Dr. Florian Knoop (now Linköping University), first author of both publications. “Such effects are typically neglected in thermal-conductivity simulations, since these defects are so short-lived and so microscopically localised compared to typical heat-transport scales, that they are assumed to be irrelevant. However, the performed calculations showed that they trigger lower thermal conductivities”, adds Dr. Christian Carbogno, a senior author of the studies. These insights may offer new opportunities to fine-tune and design thermal insulators on a nanoscale level through defect engineering, potentially contributing to advances in energy-efficient technology. Read the full publications here: Florian Knoop, Thomas A. R. Purcell, Matthias Scheffler, and Christian Carbogno Florian Knoop, Matthias Scheffler, Christian Carbogno |

SURFACES AT REALISTIC CONDITIONS
Image: images/NEWS/Phase_diagram_SI(100)_original.jpg
Figure caption: © FHI / Y. Zhou
Content:
|
Authors:
Dr. Yuanyuan Zhou
Dr. Chunye Zhu
Prof. Dr. Matthias Scheffler
|
SURFACES AT REALISTIC CONDITIONSResearchers at the NOMAD Laboratory at the Fritz Haber Institute have been engaged in describing how surfaces change in contact with reactive gas phases under different temperature and pressure conditions. For this purpose, they have developed the so-called replica exchange grand canonical method (REGC). The results were published in the journal Physical Review Letters on 17 June 2022.
_original.jpg)
© FHI / Y. Zhou
"Replica exchange" means that there are many replicas prepared for the silicon surface in contact with different hydrogen atmospheres. These replicas exchange with each other during the simulation. "Grand-canonical" means that the silicon surface in each replica exchanges deuterium atoms or molecules with the deuterium gas reservoir it touches, eventually reaching equilibrium with the deuterium gas reservoir. Knowledge of the morphology and structural evolution of material surfaces in a given reactive atmosphere is a prerequisite for understanding the mechanism of e.g. heterogeneous catalysis reactions and electrocatalysis due to the structure-property-power relationship. In general, the reliable tracking of phase equilibria is of technological importance for the reasonable design of surface properties. Read the full publication here: Y. Zhou, C. Zhu, M. Scheffler, and L. M. Ghiringhelli |


THE FAIRMAT CONCEPT PRESENTED AS A
Image: images/NEWS/FAIRmat_Concept_original.jpg
Figure caption: © Florian Knoop, NOMAD Laboratory
Content:
|
Authors
Prof. Dr. Matthias Scheffler
|
THE FAIRMAT CONCEPT PRESENTED AS A "PERSPECTIVE" IN "NATURE"Since 2020, the NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society, together with other universities and research institutes, has been developing an AI-supported research database for the field of materials science and solid-state physics. In a recent "Perspectives" article in the renowned scientific journal "Nature", the scientists involved present the concept of their data infrastructure NOMAD/FAIRmat.
"Nature" is one of the world's most-read and most prestigious multidisciplinary, academic journals. As a special component, "Nature" features "Perspectives". These are more forward looking contributions and intended to stimulate discussion and new scientific approaches. After the international Conference on a FAIR Data Infrastructure in summer 2020 the leadership of FAIRmat was invited by nature's board of editors to write a "Perspective", describing its forward looking concepts on the revolutionary impact of data-centric research in Materials Science and the Chemical Physics of Solids. The prosperity and lifestyle of our society depend to a large extent on the achievements of condensed matter physics, chemistry and materials science, because new products for energy, the environment, health, mobility, IT, etc. are largely based on improved or even novel materials. Examples include solid-state lighting, touch screens, batteries, implants, drug delivery and much more. The enormous amounts of research data produced daily in this field represent a "gold mine of the 21st century". However, this "gold mine" is of little value if these data are not comprehensively characterised and made available. This requires an efficient and easily usable research database. This is what the NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society has been developing with other research partners since 2020 with the NOMAD/FAIRmat database. The acronym FAIRdi stands for Findable, Accessible, Interoperable and Reusable Data-Infrastructure. This is intended to make it easy to share data that has already been collected and to explore it using data analysis and artificial intelligence (AI) methods. An important prerequisite for this database in the field of materials science is that the experimental (or computational) conditions and the results are actually documented in all details in such a way that the studies are reproducible and the data collection (including the comprehensive characterisation of the experimental set-up) is automated as far as possible. This sounds like an outdated requirement, but it has not yet been consistently implemented, and is essential for data-centred science. Moreover, this requires close cooperation between experts from the fields of data science, IT infrastructure, software engineering and materials science as equal partners. In FAIRmat, this is realised through a central hub of specialists at the Department of Physics at Humboldt-Universität zu Berlin. In addition, hardware for data storage and processing, advanced analyses and high-speed networks is a basic prerequisite for building the data infrastructure described. Furthermore, "middleware", e.g. for the efficient exchange of data generated in or by different digital environments, is needed. Finally, new software tools are also being developed, for example for fitting data, removing noise from data and learning the rules behind patterns in data. With such tools, it will also be possible to identify "material genes", i.e. physical parameters related to the processes that trigger, facilitate or hinder a particular material property or function. FAIRmat will promote the international coordination of such tool developments in the broader materials science community. FAIRmat will thus fill a digitisation gap in the field of materials science, as has already been done, for example, in the field of life sciences through the introduction of digital libraries. Read the full publication here: M. Scheffler, M. Aeschlimann, M. Albrecht, T. Bereau, H.-J. Bungartz, C. Felser, M. Greiner, A. Groß, C. T. Koch, K. Kremer, W. E. Nagel, M. Scheidgen, C. Wöll, and C. Draxl,
|

TURNING GREENHOUSE GASES INTO USEFUL CHEMICALS
Image: images/NEWS/Useful_chemicals_original.jpg
Figure caption: © C. Arndt-Sullivan
Content:
|
Authors
Dr. Aliaksei Mazheika
Y. Wang
|
TURNING GREENHOUSE GASES INTO USEFUL CHEMICALSCO2 is a critical pollutant of the atmosphere with noticeable climatic consequences. However, routes already exist to convert CO2 into useful chemicals or fuels. The key is catalysis – a process of accelerating desired chemical reactions, involving special materials (catalysts).
© C. Arndt-Sullivan
But the known catalysts are too inefficient for making this conversion practicable. An important step is the identification of materials that “activate” CO2, i.e., make the molecule ready for a chemical reaction. Using high-throughput calculations and subgroup discovery, scientists at the NOMAD Laboratory identified the materials properties (genes) and rules that classify potentially good catalytic materials for the desired purpose. Because of its chemical stability, CO2 is presently one of the critical mankind-created greenhouses gases. However, at some point CO2 may well become a raw material for creating fuels and valuable chemicals. The catalytic, chemical conversion towards methane (viable for combustion engines and heating) and other important chemicals is all possible, already today, but the process is very inefficient. We need better catalysts. The NOMAD Laboratory developed and advanced artificial intelligence (AI) methods that enable the identification of basic materials parameters that correlate with materials properties and functions of interest (here the activation of CO2). These parameters are also called materials genes as they correlate with different mechanisms that trigger, but maybe just facilitate or even hinder the different processes playing together, depending on their combination – very much as genes in biology. In the coordinate system of these genes, regions are identified where good catalytic materials can be found. Specifically, this CO2 study employed the AI method called “subgroup discovery”, and it focused on the wide class of metal oxides. Catalysis happens at surfaces. Thus, the study also considered various surfaces of all these materials. Altogether, 141 different surfaces where calculated (71 different materials) with state-of-the-art density-functional-theory high-throughput methodology. The results were then used for training the AI. In general terms, this study also represents a conceptual change of modeling heterogeneous catalysis (and other materials functions). In the past it was attempted to calculate the full catalytic process. However, it became clear that the many aspects that rule heterogeneous catalysis, e.g. the dynamical restructuring of the surface under reaction conditions, are too intricate, and that a full theoretical description does not make sense. Thus, a combined approach linking high-throughput calculations, AI, and experimental results appears more appropriate and was suggested in this study. Read the full publication here: A. Mazheika, Y. Wang, R. Valero, F. Vines, F. Illas, L. Ghiringhelli, S. Levchenko, and M. Scheffler |
